My first scientific paper: PMut reinvented
7 Sep, 2017
Last July, my first scientific paper was published. Titled PMut. A web-based tool for the annotation of pathological variants on proteins, 2017 update, it is the result of more than two years work in my Ph.D. in bioinformatics. The paper has been published in the English journal Nucleic Acids Research, one of the main journals in the biomedicine field; specifically, it is part of the yearly web services special issue.
In this blog article, I will try to summarize the most relevant points of the paper, ommiting the technical details. If you have not read the article "Introduction to my research in bioinformatics", I recommend it as an introduction to the context of my research.
Classic PMut
PMut is a predictor of the pathology of protein mutations. That is, it is a computer program that, given a mutation in a protein, gives us a prediction of whether this mutation will cause a disease or it will be harmless.
This idea is not new, in fact the PMut predictor was created in 2005 and has been one of the most widely used predictors since then. However, during this last decade, the predictive methods have evolved dramatically and the number of mutations analyzed in the lab has increased by a factor of 10. Thus, I took the challenge of updating this predictor, and this scientific paper is the first result of my work.
The PyMut prediction engine
As a prelude to the development of a new predictor, we have developed the PyMut prediction engine in the form of a Python package. This software, publicly available, is capable of managing all the tasks related to pathology predictions: the calculation of numerical descriptors of mutations, the training of predictors, the evaluation of these, the generation of related charts, etc. Here are some examples of the charts we can get using PyMut:
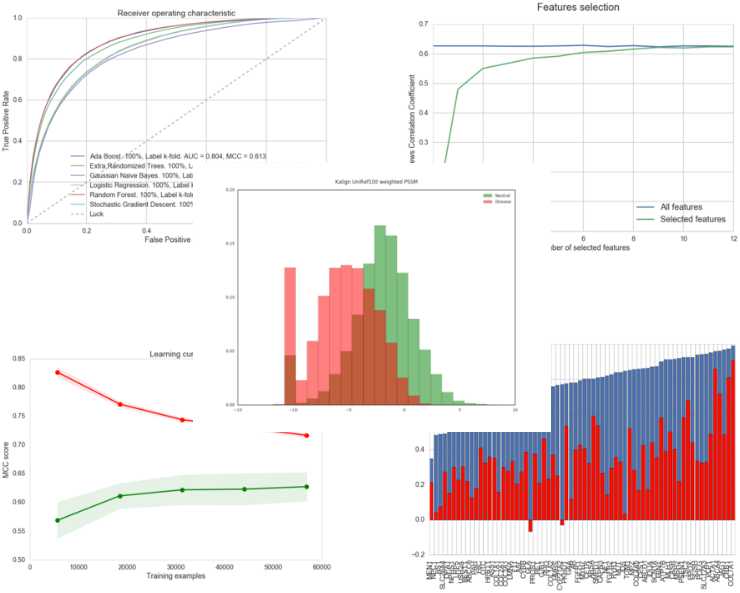 Examples of charts generated using PyMut.
Examples of charts generated using PyMut.
With the PyMut package, everyone can replicate on their computer the process of preparing, testing and using a predictor such as PMut.
The new PMut2017 predictor
Using the PyMut engine, and training with the SwissVar database, which contains 27,203 pathogenic mutations and 38,078 neutral mutations over more than 12,000 different proteins, we have trained the PMut2017 predictor.
Before giving the predictor for good, it is crucial to evaluate its reliability. In fact, the journal demands a very strict study of its precision, and for this reason we have followed 4 different approaches:
- 10-fold cross-validation. It consists of dividing the training data into 10 sets, training a predictor with 9 of them and evaluating with the one that has been excluded. We repeat this procedure 10 times, excluding a different set every time and averaging the results. This is the easiest way to obtain an approximation of the reliability of the predictor.
- Blind test using new SwissVar data. We move to December 2015, and train a predictor with the data that was available back then in SwissVar. Then, we evaluate the 3,166 mutations that were added to SwissVar during 2016.
- ClinVar data. Clinvar is another database of annotated mutations, significantly different from SwissVar. We have evaluated how good are the predictions of PMut2017 on the Clinvar mutations that are not present in SwissVar.
- Study of specific genes. We compared how PMut2017 works for some specific genes, comparing the results with some of the most widely used predictors.
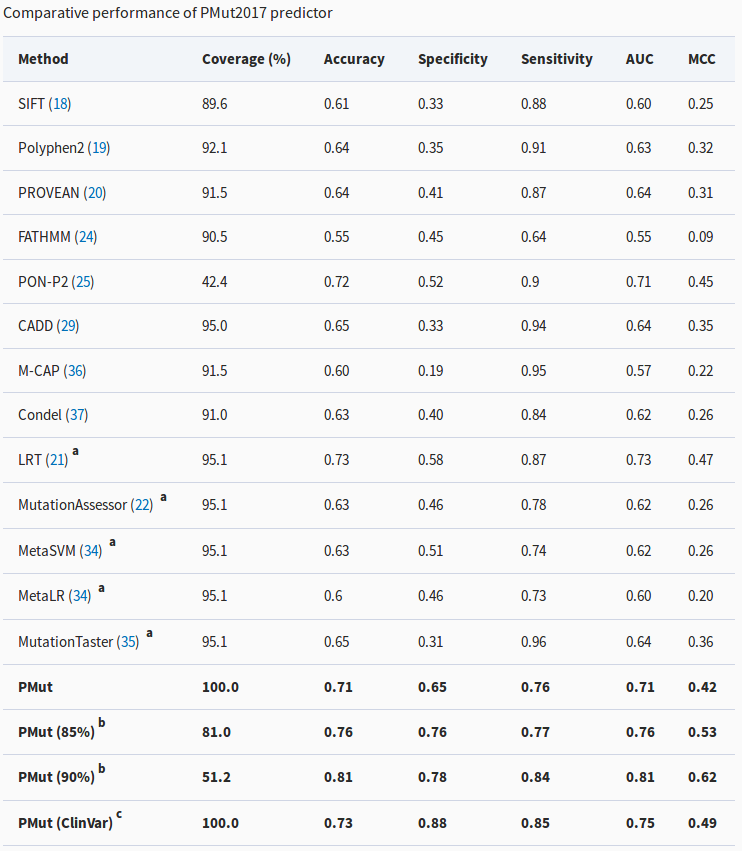 Comparison of PMut with other predictors. The most relevant metric is the Matthews Correlations Coefficient (MCC, last column), for which a higher number means better predictions. See the article to understand the details of the table.
Comparison of PMut with other predictors. The most relevant metric is the Matthews Correlations Coefficient (MCC, last column), for which a higher number means better predictions. See the article to understand the details of the table.
All these assessments have allowed us to compare PMut2017 with the most accurate predictors and to verify that it is undoubtedly among the most powerful ones.
The PMut web portal
To make predictions accessible to the research community, we facilitate the use of PMut2017 from a web page open to everyone. Users can send a list of mutations through a form and obtain the corresponding predictions.
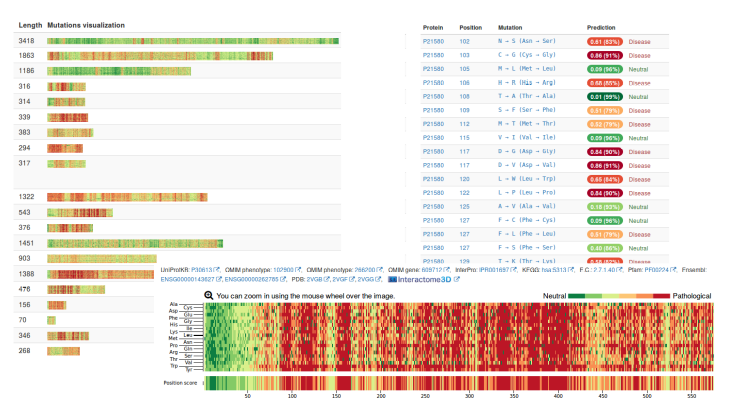 Screenshots of the web portal.
Screenshots of the web portal.
The vast majority of users ask for the impact of mutations on human proteins. In order to streamline these calculations, and since I do the Ph.D. in the Barcelona Supercomputing Center and we like to use the enormous supercomputer we have, we decided to pre-calculate some predictions. In fact not only some, but all the possible mutations in all human proteins, that is, a total of 725,596,928 mutations over 106,407 proteins. These predictions, stored in a MongoDB database, are immediately accessible without the need for any calculation.
 Marenostrum 4, BSC's supercomputer, has a computing power of 11.1 Petaflops, that is to say, 11.1 × 1015 operations per second. Marenostrum 4 was released in July 2017; for my computations I used the late Marenostrum 3.
Marenostrum 4, BSC's supercomputer, has a computing power of 11.1 Petaflops, that is to say, 11.1 × 1015 operations per second. Marenostrum 4 was released in July 2017; for my computations I used the late Marenostrum 3.
The PMut portal also offers the novel possibility of training specific predictors. This functionality is especially relevant to those researchers that work on one or more specific proteins. These researchers, who usually have more data than the public, can easily train a custom predictor and use it for their specific research.
Conclusion
In summary, in this work we have published a new predictor as accurate as the best predictors currently available. In addition, we share the code we used to train it, released as a Python package. Finally, we make all these functions accessible from a web portal where, for the first time, researchers can train —in a fully automatic way— tailor-made predictors for their studies.